
Annotate a VCF
Annotate your VCF file
In this post, we will construct a custom tool to parse and annotate a VCF file (v4.1), a common file in genomics. Variants will be annotated via the ExAC database API. I recommend running the script in a virtual environment (I used Anaconda for this), and creating a YAML file with the dependencies listed, to ensure portability and reproducability.
Description of a VCF file
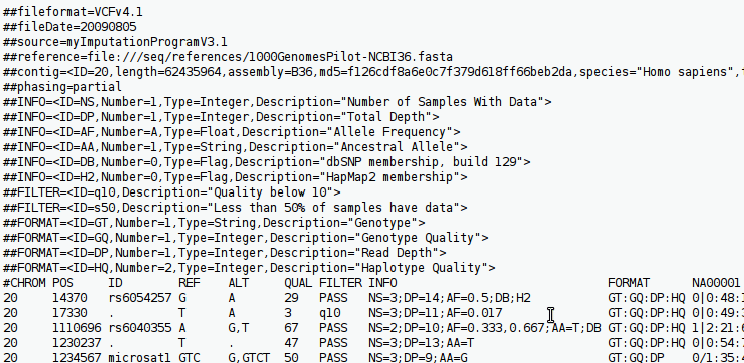
A VCF file has three sections:
-
Each meta-data line in VCF begins with ‘##’
-
Then there is a single header line beginning with a single hash (‘#’) character.
-
Data & Sample
After the header line there are data lines, with each data line describing a genetic variant at a particular position relative to the reference genome of whichever species you are studying.
Each data line is divided into tab-separated fields. There are nine fixed fields: “CHROM”, “POS”, “ID”, “REF”, “ALT”, “QUAL”, “FILTER”, “INFO” and “FORMAT”.
The sample fields follow the fixed fields, usually with a genotype call and related data.
Import required libraries
import json
import pandas as pd
import re
import sys
import time
import urllib.request
Don’t forget to set the important, hard-coded variables:
- ExAC API
- Ensembl list of variant effect severity.
ExAC API:
‘http://exac.hms.harvard.edu/rest/bulk/variant’
Ensembl variant consequences
Ensembl variation effect, in order of severity (descending):
effect_ranked = [
'transcript_ablation',
'splice_acceptor_variant',
'splice_donor_variant',
'stop_gained',
'frameshift_variant',
'stop_lost',
'start_lost',
'transcript_amplification',
'inframe_insertion',
'inframe_deletion',
'missense_variant',
'protein_altering_variant',
'splice_region_variant',
'incomplete_terminal_codon_variant',
'stop_retained_variant',
'synonymous_variant',
'coding_sequence_variant',
'mature_miRNA_variant',
'5_prime_UTR_variant',
'3_prime_UTR_variant',
'non_coding_transcript_exon_variant',
'intron_variant',
'NMD_transcript_variant',
'non_coding_transcript_variant',
'upstream_gene_variant',
'downstream_gene_variant',
'TFBS_ablation',
'TFBS_amplification',
'TF_binding_site_variant',
'regulatory_region_ablation',
'regulatory_region_amplification',
'feature_elongation',
'regulatory_region_variant',
'feature_truncation',
'intergenic_variant'
]
Create the class to parse the VCF
class VCF_Reader(object):
'''
A class to parse a VCF file
Description of a VCF file
A VCF file has three sections.
1) Each META-DATA line in VCF begins with '##'
2) Then there is a single header line beginning with a single hash (‘#’) character.
Data & Sample
3) After the header line there are DATA lines,
with each data line describing a genetic variant at a particular position relative to the reference genome
of whichever species you are studying.
Each data line is divided into tab-separated fields.
There are nine fixed fields:
"CHROM", "POS", "ID", "REF", "ALT", "QUAL", "FILTER", "INFO" and "FORMAT".
The SAMPLE fields follow the fixed fields, usually with a genotype call and related data.
'''
def __init__(self, in_vcf):
'''
Create VCF_Reader object
Args:
in_vcf: the VCF file to read. Required.
'''
vcf = []
file = open(in_vcf, 'r')
# Process line by line
for line in file:
if line.startswith('#CHROM'):
data_cols = line.strip('\n').split('\t')
sample_data = re.findall('FORMAT\t(.+)', line)[0].split('\t')
feat_header = ['variant_type']
for sample in sample_data:
feat_header += [sample + '_depth', sample + '_num_reads', sample + '_percent_variant_support']
feat_header += ['exac_allele_freq']
if line[0] != '#':
row = line.rstrip('\n').split('\t')
vcf.append(row)
file.close()
self.keys = data_cols
self.vcf = vcf
self.samples = sample_data
self.header_cols = feat_header
def get_type(self):
'''
Extracts the variant type from the VCF file for each variant
'''
type_var = []
for variant in self.vcf:
# Combine keys with variants in dict
var_dict = dict(zip(self.keys, variant))
var_info = var_dict["INFO"]
# Find variant types under INFO
var_anno = re.findall('TYPE=(.+)', var_info)
type_var.append(var_anno)
return type_var
def get_seq_depth(self):
'''
Returns for each sample and variant: the read depth, alt. read depth, and alt. AF.
'''
s_AD = []
for entry in self.vcf:
var_dict = dict(zip(self.keys, entry))
AD = []
for i in range(len(self.samples)):
info_s = var_dict[self.samples[i]]
al_depth = int(info_s.split(':')[3].split(',')[0]) # Third entry contains counts per allele
al_counts = int(info_s.split(':')[3].split(',')[1])
AF = al_counts/al_depth
AD += [al_depth, al_counts, AF]
s_AD.append(AD)
return s_AD
def get_var_ids(self):
'''
Returns a variant ID composed of 'chromosome-position-reference-alternative' which is used in performing ExAC queries
'''
var_ids = []
for variant in self.vcf:
var_dict = dict(zip(self.keys, variant))
info_v = '-'.join([var_dict['#CHROM'],
var_dict['POS'],
var_dict['REF'],
var_dict['ALT']])
var_ids.append(info_v)
return var_ids
Create the class to call the ExAC API
An Internet connection is required to connect to the database.
class api_query(object):
'''
A class to query variant IDs in ExAC. For each variant, append its allele frequency and genotype consequence(s).
Example variant ID: 14-21853913-T-C
Column headers: chromosome #, position, reference allele, alternative allele.
Field separation: "-"
Args:
var_ids: the variant IDs to query. Derived from the VCF_Reader class get_var_ids(). Required.
'''
def __init__(self,var_ids):
exac_url = 'http://exac.hms.harvard.edu/rest/bulk/variant'
ids = json.dumps(var_ids)
ids = ids.encode('ASCII')
# Establish connection with ExAC API
time.sleep(0.3)
api_call = urllib.request.Request(exac_url, data=ids, method='POST')
exac_query = urllib.request.urlopen(api_call)
exac_data = json.loads(exac_query.read().decode())
self.var_ids = var_ids
self.query_res = exac_data
self.num_rows = len(var_ids)
def get_AF(self):
'''
On success: returns allele frequency (AF)
'''
AF = []
for variant in range(self.num_rows):
var = self.query_res[self.var_ids[variant]]['variant']
if 'allele_freq' in var.keys():
AF.append([var['allele_freq']])
else:
AF.append([float("nan")])
return AF
def get_csqs(self):
'''
On success: returns most deleterious consequence from ExAC.
'''
csqs = []
for i in range(self.num_rows):
v = self.query_res[self.var_ids[i]]
if v['consequence']:
cq_list = list(v['consequence'].keys())
ranked_sev = [] = []
for c in cq_list:
if c in effect_ranked:
ranked_sev.append(effect_ranked.index(c))
else:
ranked_sev.append(float("inf"))
# Get consequence of maximum severity in the pair: (index, csqs)
index = min([(index, csqs) for (csqs, index) in enumerate(ranked_sev)])[1]
csqs.append(cq_list[index])
else:
# Return NA if no consequence found.
csqs.append('NA')
return csqs
Create the main function
Use pandas to output a CSV file.
def main(infile, outfile):
'''
Write annotated VCF to CSV file.
'''
print("Parsing VCF file!")
start = time.time()
data = VCF_Reader(in_vcf = infile)
variant_type = data.get_type()
variant_depth = data.get_seq_depth()
var_ids = data.get_var_ids()
print('Connecting to ExAC: Querying variants!')
exac_anno = api_query(var_ids)
# Get allele frequency
AF = exac_anno.get_AF()
# Get allele consequence
csqs = exac_anno.get_csqs()
# Concatenate features
features = []
for var in range(len(variant_type)):
if csqs[var]:
variant_type[var][0] += ' (VEP: ' + csqs[var] + ')'
features.append(
variant_type[var]
+ variant_depth[var]
+ AF[var]
)
print("Writing to CSV: " + sys.argv[2])
features_df = pd.DataFrame(features, columns=data.header_cols)
# Create CSV file
features_df.to_csv(outfile, sep='\t', header=True, index=False)
print("Done! Annotated {} variants in {}.".format(len(var_ids), time.time() - start))
Read in command line arguments
if __name__ == "__main__":
# Parse command line args
if len(sys.argv) == 3:
main(infile = sys.argv[1], outfile = sys.argv[2])
print("vcf_parser called directly.")
else:
print("vcf_parser imported into another module.")
Conclusion
Given a VCF file (v4.1), this script should output a CSV file with ExAC annotations.
